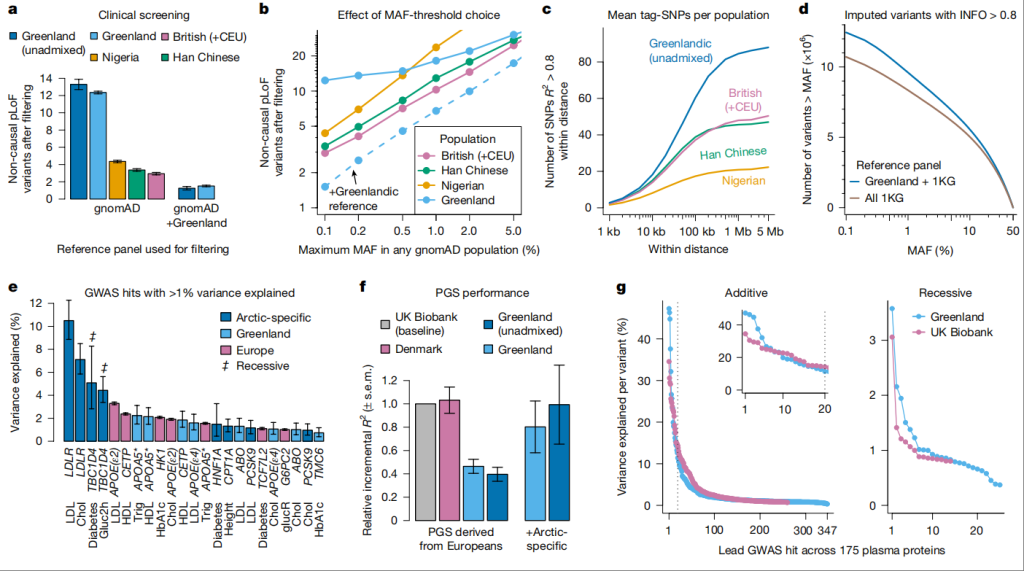
图2遗传架构对疾病定位的影响
a,在参考面板(gnomAD v.3.0.0)中移除任何群体中MAF>0.1%的变异后,剩余的非因果pLoF变异的平均数量(标准误)。
b,与a相同,但在参考面板中使用不同的MAF阈值。c,标记SNP(R²>0.8)的平均数量作为焦点SNP距离的函数。
d,INFO评分大于0.8且MAF超过x轴所示阈值的推断变异数量。推断使用格陵兰全基因组测序合并1KG参考面板(n=4483)或仅使用1KG参考面板(n=3202)进行。
e,f,格陵兰和欧洲13项代谢性状最大全基因组关联分析(GWAS)的比较。
e,在格陵兰和欧洲13项代谢性状最大GWAS中,解释超过1%方差的全基因组关联(95%置信区间)。基因名位于条形图下方,与变异相关的表型列在基因名下方。对于糖尿病,我们使用了责任尺度方差解释。星号表示该区域中的因果基因不确定。Chol,总胆固醇;Gluc2h,葡萄糖(2小时);GlucR,葡萄糖(随机);HbA1c,血红蛋白A1c;HDL,高密度脂蛋白;Trig,甘油三酯。
f,欧洲衍生的多基因评分(PGS)预测相应13项代谢性状(归一化至英国生物银行)的平均增量R²(标准误),针对所有格陵兰参与者、仅未混合的格陵兰参与者或丹麦参与者。右侧的两个条形图是添加北极特异性变异后的平均增量R²。
g,格陵兰和英国生物库中175种血浆蛋白(Olink)全基因组显著关联的领先SNP解释的方差,按解释的方差排序,并按产生最低P值的模型分组;n=3707。插图显示了前20个GWAS hits的放大图。